Peptide Search¶
In MS-based proteomics, fragment ion spectra (MS2 spectra) are often interpreted by comparing them against a theoretical set of spectra generated from a FASTA database. OpenMS contains a (simple) implementation of such a “search engine” that compares experimental spectra against theoretical spectra generated from an enzymatic or chemical digest of a proteome (e.g. tryptic digest).
SimpleSearch¶
In most proteomics applications, a dedicated search engine (such as Comet,
Crux, Mascot, MSGFPlus, MSFragger, Myrimatch, OMSSA, SpectraST or XTandem;
all of which are supported by pyOpenMS) will be used to search data. Here, we will
use the internal SimpleSearchEngineAlgorithm from OpenMS used for teaching
purposes. This makes it very easy to search an (experimental) mzML file against
a fasta database of protein sequences:
from urllib.request import urlretrieve
# from urllib import urlretrieve # use this code for Python 2.x
from pyopenms import *
gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop"
urlretrieve (gh +"/src/tests/topp/SimpleSearchEngine_1.mzML", "searchfile.mzML")
urlretrieve (gh +"/src/tests/topp/SimpleSearchEngine_1.fasta", "search.fasta")
protein_ids = []
peptide_ids = []
SimpleSearchEngineAlgorithm().search("searchfile.mzML", "search.fasta", protein_ids, peptide_ids)
This will print search engine output including the number of peptides and proteins in the database and how many spectra were matched to peptides and proteins:
Peptide statistics
unmatched : 0 (0 %)
target/decoy:
match to target DB only: 2 (100 %)
match to decoy DB only : 0 (0 %)
match to both : 0 (0 %)
PSM inspection¶
We can now investigate the individual hits as we have done before in the Identification tutorial.
for peptide_id in peptide_ids:
# Peptide identification values
print (35*"=")
print ("Peptide ID m/z:", peptide_id.getMZ())
print ("Peptide ID rt:", peptide_id.getRT())
print ("Peptide scan index:", peptide_id.getMetaValue("scan_index"))
print ("Peptide scan name:", peptide_id.getMetaValue("scan_index"))
print ("Peptide ID score type:", peptide_id.getScoreType())
# PeptideHits
for hit in peptide_id.getHits():
print(" - Peptide hit rank:", hit.getRank())
print(" - Peptide hit charge:", hit.getCharge())
print(" - Peptide hit sequence:", hit.getSequence())
mz = hit.getSequence().getMonoWeight(Residue.ResidueType.Full, hit.getCharge()) / hit.getCharge()
print(" - Peptide hit monoisotopic m/z:", mz)
print(" - Peptide ppm error:", abs(mz - peptide_id.getMZ())/mz *10**6 )
print(" - Peptide hit score:", hit.getScore())
We notice that the second peptide spectrum match (PSM) was found for the third
spectrum in the file for a precursor at 775.38 m/z for the sequence
RPGADSDIGGFGGLFDLAQAGFR.
tsg = TheoreticalSpectrumGenerator()
thspec = MSSpectrum()
p = Param()
p.setValue("add_metainfo", "true")
tsg.setParameters(p)
peptide = AASequence.fromString("RPGADSDIGGFGGLFDLAQAGFR")
tsg.getSpectrum(thspec, peptide, 1, 1)
# Iterate over annotated ions and their masses
for ion, peak in zip(thspec.getStringDataArrays()[0], thspec):
print(ion, peak.getMZ())
e = MSExperiment()
MzMLFile().load("searchfile.mzML", e)
print ("Spectrum native id", e[2].getNativeID() )
mz,i = e[2].get_peaks()
peaks = [(mz,i) for mz,i in zip(mz,i) if i > 1500 and mz > 300]
for peak in peaks:
print (peak[0], "mz", peak[1], "int")
Comparing the theoretical spectrum and the experimental spectrum for
RPGADSDIGGFGGLFDLAQAGFR we can easily see that the most abundant ions in the
spectrum are y8 (877.452 m/z), b10 (926.432), y9 (1024.522 m/z) and b13
(1187.544 m/z).
Visualization¶
When loading the searchfile.mzML into the OpenMS
visualization software TOPPView, we can convince ourselves that the observed
spectrum indeed was generated by the peptide RPGADSDIGGFGGLFDLAQAGFR by loading
the corresponding theoretical spectrum into the viewer using “Tools”->”Generate
theoretical spectrum”:
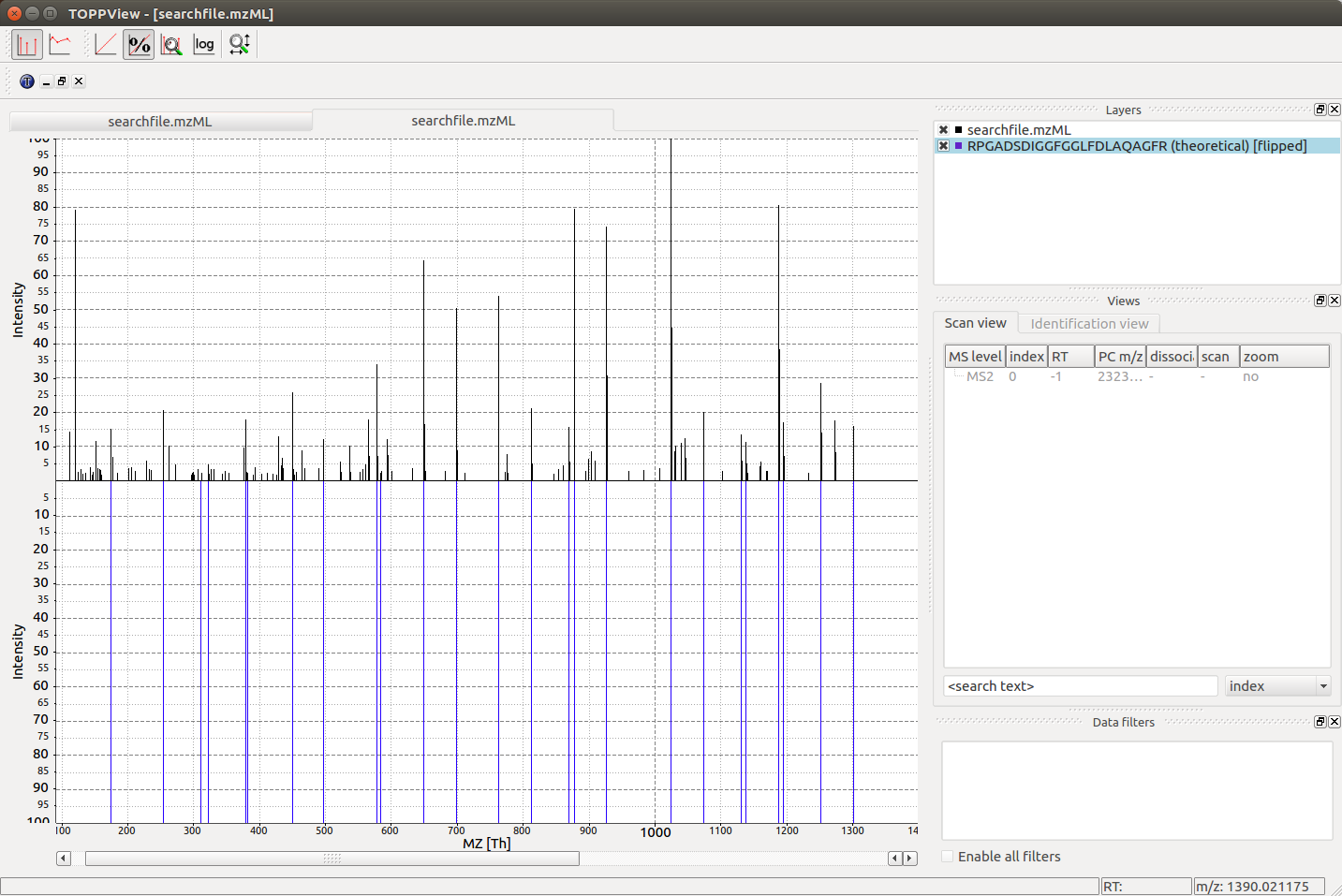
From our output above, we notice that the second peptide spectrum match (PSM)
at 775.38 m/z for sequence RPGADSDIGGFGGLFDLAQAGFR was found with an error
tolerance of 2.25 ppm, therefore if we set the precursor mass tolerance to 4
ppm (+/- 2ppm), we expect that we will not find the hit at 775.38 m/z any more:
salgo = SimpleSearchEngineAlgorithm()
p = salgo.getDefaults()
print ( p.items() )
p[b'precursor:mass_tolerance'] = 4.0
salgo.setParameters(p)
protein_ids = []
peptide_ids = []
salgo.search("searchfile.mzML", "search.fasta", protein_ids, peptide_ids)
print("Found", len(peptide_ids), "peptides")
As we can see, using a smaller precursor mass tolerance leads the algorithm to
find only one hit instead of two. Similarly, if we use the wrong enzyme for
the digestion (e.g. p[b'enzyme'] = "Formic_acid"), we find no results.