
Interfacing with ML Libraries#
Overview#
Machine Learning is the field of study that gives computers the capability to learn without being explicitly programmed. Machine learning (ML) is well known for its powerful ability to recognize patterns and signals. Recently, the mass spectrometry community has embraced ML techniques for large-scale data analysis.
Predicting accurate retention times (RT) has shown to improve identification in bottom-up proteomics.
In this tutorial we will predict the RT from amino acid sequence data using simple machine learning methods.
First, we import all necessary libraries for this tutorial.
1!pip install seaborn
2!pip install xgboost
1import pandas as pd
2import seaborn as sns
3import matplotlib.pyplot as plt
4import string
5from collections import Counter
6import numpy as np
7from urllib.request import urlretrieve
Once we have imported all libraries successfully, we are going to store the dataset in a variable.
1gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop/doc/pyopenms"
2urlretrieve(gh + "/src/data/pyOpenMS_ML_Tutorial.tsv", "data.tsv")
3tsv_data = pd.read_csv("data.tsv", sep="\t", skiprows=17)
Here, we have prepared a tsv file that contains three columns sequence, RT and charge.
Note that this table could also be easily created from identification data as produced in previous chapters.
Before we move forward lets try to understand more about our data:
a. Sequence - Chains of amino acids form peptides or proteins. The arrangement of amino acids is referred as amino acid sequence. The composition and order of amino acids affect the physicochemical properties of the peptide and lead to different retention in the column. b. Retention time (RT) - is the time taken for an analyte to pass through a chromatography column.
From the amino acid sequence we can derive additional properties (machine learning features) used to train our model.
We can easily check for its shape by using the tsv_data.shape attribute, which will return the size of the dataset.
1print(tsv_data.shape)
(15896, 3)
Explore the top 5 rows of the dataset by using head() method on pandas DataFrame.
1tsv_data.head()
sequence RT charge
0 EEETVAK 399.677766 2
1 EQEEQQQQEGHNNK 624.555300 3
2 SHGGHTVISK 625.797960 3
3 SGTHNMYK 625.982520 2
4 AARPTRPDK 626.073300 3
As the RT column is our response variable, we will be storing it separately as Y1_test
1Y1_test = tsv_data["RT"]
Preprocessing#
Cleaning data before applying a machine learning method keeps the relevant information in potentially massive amount of data.
Here we will apply some simple preprocessing to extract novel machine learning features from the amino acid sequences. Some of the parameters that can be derived are
{Alphabet}_count = The count of Amino Acids in the sequence.
{Alphabet}_freq = The count of Amino Acids divided by the total length of the sequence.
length = The total number of amino acids in the sequence.
1alphabet_list = list(string.ascii_uppercase)
2column_headers = (
3 ["sequence"]
4 + [i + "_count" for i in alphabet_list]
5 + [i + "_freq" for i in alphabet_list]
6 + ["charge", "length"]
7)
8types = (
9 ["object"]
10 + ["int64" for i in alphabet_list]
11 + ["float64" for i in alphabet_list]
12 + ["int64", "int64"]
13)
14pdcols = dict(zip(column_headers, types))
As we have all the column names, now we will start populating it.
1df = pd.DataFrame(
2 np.zeros((len(tsv_data.index), len(column_headers))), columns=column_headers
3)
4
5df["sequence"] = tsv_data["sequence"]
6df["charge"] = tsv_data["charge"]
7
8# For populating the length column
9df["length"] = df["sequence"].str.len()
10
11df = df.astype(dtype=pdcols)
12
13
14# For populating the {alphabet}_count columns
15def count(row):
16 counts = Counter(row["sequence"])
17 for count in counts:
18 row[count + "_count"] = int(counts[count])
19 return row
20
21
22df = df.apply(lambda row: count(row), axis=1)
23df.head()
sequence A_count B_count C_count D_count E_count F_count G_count H_count I_count ... S_freq T_freq U_freq V_freq W_freq X_freq Y_freq Z_freq charge length
0 EEETVAK 1 0 0 0 3 0 0 0 0 ... 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 2 7
1 EQEEQQQQEGHNNK 0 0 0 0 4 0 1 1 0 ... 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 3 14
2 SHGGHTVISK 0 0 0 0 0 0 2 2 1 ... 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 3 10
3 SGTHNMYK 0 0 0 0 0 0 1 1 0 ... 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 2 8
4 AARPTRPDK 2 0 0 1 0 0 0 0 0 ... 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 3 9
Now we have completed all the data preprocessing steps. We have deduced a good amount of information from the amino acid sequences that might have influence on the retention time in the column.
Now we are good to proceed on building the machine learning model.
Modelling#
1import seaborn as sns
2import matplotlib.pyplot as plt
3
4from sklearn.model_selection import StratifiedKFold
5from xgboost import XGBRegressor
6from sklearn.model_selection import train_test_split
7from matplotlib import pyplot
8from sklearn.metrics import mean_squared_error
9from sklearn.model_selection import ShuffleSplit
1test_df = df.copy()
2test_df = test_df.drop("sequence", axis=1)
Now, we create the train and test set for cross-validation of the results
using the train_test_split function from sklearn’s model_selection module with test_size
size equal to 30% of the data. To maintain reproducibility of the results, a random_state is also assigned.
1# Splitting Test data into test and validation
2X_train, X_test, Y_train, Y_test = train_test_split(
3 test_df, Y1_test, test_size=0.3, random_state=3
4)
We will be using the XGBRegressor() class because it is clearly a regression problem as the response variable ( retention time ) is continuous.
1xg_reg = XGBRegressor(
2 n_estimators=300,
3 random_state=3,
4 max_leaves=5,
5 colsample_bytree=0.7,
6 max_depth=7,
7)
Fit the regressor to the training set and make predictions on the test set using the familiar .fit() and .predict() methods.
1xg_reg.fit(X_train, Y_train)
2Y_pred = xg_reg.predict(X_test)
Compute the root mean square error (rmse) using the mean_sqaured_error function from sklearn’s metrics module.
1rmse = np.sqrt(mean_squared_error(Y_test, Y_pred))
2print("RMSE: %f" % (rmse))
RMSE: 437.017290
Store the Observed v/s Predicted value in pandas dataframe and print.
1k = pd.DataFrame(
2 {"Observed": Y_test.values.flatten(), "Predicted": Y_pred.flatten()}
3)
4print(k)
Observed Predicted
0 3652.28442 3927.141846
1 4244.80320 4290.294434
2 3065.19054 3703.156982
3 909.50610 762.218567
4 1982.80902 2628.958740
... ... ...
4764 5527.23804 5599.530762
4765 3388.76430 3272.557617
4766 3101.35566 3346.364990
4767 5515.94682 5491.597168
4768 2257.63092 2258.312988
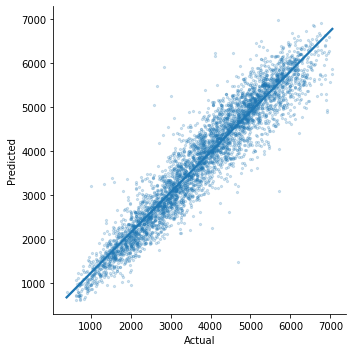
We will now generate a Observed v/s Predicted plot that gives a high level overview about the model performance. We can clearly see that only few outliers are there and most of them lie in between the central axis. This means that prediction actually works and observed and predicted value won’t differ too much.
1sns.lmplot(
2 x="Observed", y="Predicted", data=k, scatter_kws={"alpha": 0.2, "s": 5}
3)
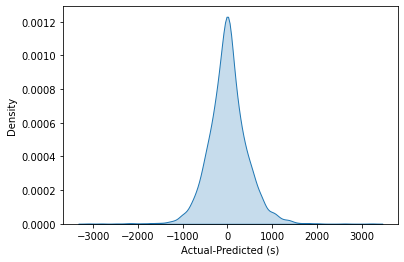
1p = sns.kdeplot(data=k["Observed"] - k["Predicted"], fill=True)
2p.set(xlabel="Observed-Predicted (s)")
In order to build more robust models, it is common to do a k-fold cross validation where all the entries in the original training dataset are used for both training as well as validation. Also, each entry is used for validation just once. XGBoost supports k-fold cross validation via the cv() method. All we have to do is specify the nfolds parameter, which is the number of cross validation sets we want to build.
1# Performing k-fold cross validation
2X = np.arange(10)
3ss = ShuffleSplit(n_splits=5, test_size=0.25, random_state=0)
4performance_df = pd.DataFrame()
5performance_list = []
6counter = 0
7for train_index, test_index in ss.split(X_train, Y_train):
8 counter += 1
9
10 X_train_Kfold, X_test_Kfold = (
11 X_train[X_train.index.isin(train_index)].to_numpy(),
12 X_train[X_train.index.isin(test_index)].to_numpy(),
13 )
14 y_train_Kfold, y_test_Kfold = (
15 Y_train[Y_train.index.isin(train_index)].to_numpy().flatten(),
16 Y_train[Y_train.index.isin(test_index)].to_numpy().flatten(),
17 )
18
19 Regressor = XGBRegressor()
20 Regressor.fit(X_train_Kfold, y_train_Kfold)
21
22 predictions = Regressor.predict(X_test_Kfold)
23
24 df = pd.DataFrame(
25 {"Observed": y_test_Kfold.flatten(), "Predicted": predictions.flatten()}
26 )
27
28 print("Fold-" + str(counter))
29 print("---------------------")
30 print(df)
Fold-1
---------------------
Observed Predicted
0 1845.17346 2051.894043
1 1155.68124 1911.122192
2 2847.94272 2753.223145
3 2370.70494 2670.160889
4 4111.31718 3961.675049
... ... ...
1935 3880.18458 3454.832031
1936 4125.82776 4068.806152
1937 4586.33838 3829.927002
1938 2261.99454 3225.578613
1939 4342.82430 3943.912354
[1940 rows x 2 columns]
Fold-2
---------------------
Observed Predicted
0 3476.56062 4075.536377
1 4009.78704 4022.654785
2 2847.94272 2779.675293
3 3669.33108 4026.944824
4 3997.12632 3566.471436
... ... ...
1907 2916.91818 2744.992676
1908 3569.64318 3862.661621
1909 2118.25278 2221.599854
1910 1787.61012 1839.471802
1911 3583.44846 3210.243164
[1912 rows x 2 columns]
Fold-3
---------------------
Observed Predicted
0 2052.18066 2237.868896
1 4336.45050 3622.901367
2 2317.39104 2496.773438
3 3356.40018 3291.187988
4 1778.73198 2034.299683
... ... ...
1934 3795.23424 2968.955322
1935 3622.34358 3203.385742
1936 2261.99454 3115.011475
1937 4112.62578 3743.435791
1938 4342.82430 3721.162842
[1939 rows x 2 columns]
Fold-4
---------------------
Observed Predicted
0 1762.89840 1691.997803
1 1292.39622 1418.658325
2 1914.00468 1779.962769
3 4571.86566 4618.782715
4 2317.39104 2417.823242
... ... ...
1985 2779.37664 2702.244385
1986 4335.23442 3733.191162
1987 2916.91818 2609.322021
1988 4125.82776 3947.512939
1989 3429.54294 3550.206787
[1990 rows x 2 columns]
Fold-5
---------------------
Observed Predicted
0 2790.00414 3010.381592
1 3476.56062 3972.215820
2 1845.17346 1901.611572
3 4009.78704 3884.857178
4 3578.05344 2993.831787
... ... ...
1975 3778.69704 4209.392090
1976 1494.22332 1612.613281
1977 4125.82776 3902.622559
1978 4701.03624 4372.867676
1979 1888.41552 2342.040771
[1980 rows x 2 columns]
That’s it, we trained a simple machine learning model to predict peptide retention times from peptide data.
Sophisticated machine models integrate retention time data from many experiments add additional properties (or even learn them from data) of peptides to achieve lower prediction errors.