Feature Detection#
One very common task in mass spectrometry is the detection of 2-dimensional patterns in m/z and time (RT) dimension from a series of MS1 scans. These patterns are called a term:Feature and they exhibit a chromatographic elution profile in the time dimension and an isotopic pattern in the m/z dimension (see previous section for the 1-dimensional problem).
OpenMS has multiple tools that can identify these features in 2-dimensional
data, these tools are called FeatureFinder. Currently the following
FeatureFinders are available in pyOpenMS:
FeatureFinderAlgorithmPicked(Label-free, identification free feature detection of peptides)
FeatureFinderIdentificationAlgorithm(Label-free identification-guided feature detection of peptides)
FeatureFinderMultiplexAlgorithm(e.g., SILAC, Dimethyl labeling, (and label-free), identification free feature detection of peptides)
FeatureFindingMetabo(Label-free, identification free feature detection of metabolites)
FeatureFinderAlgorithmMetaboIdent(Label-free, identification guided feature detection of metabolites)
All of the algorithms above are for proteomics data with the exception of FeatureFindingMetabo and FeatureFinderAlgorithmMetaboIdent for metabolomics data and small molecules in general.
Proteomics#
Two of the most commonly used feature finders for proteomics in OpenMS are the FeatureFinderAlgorithmPicked, FeatureFinderMultiplexAlgorithm and FeatureFinderIdentificationAlgorithm which all work on (high
resolution) centroided data (FeatureFinderMultiplexAlgorithm can also work on profile data). We can use the following code to find features in MS data:
from urllib.request import urlretrieve
import pyopenms as oms
gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop/doc/pyopenms"
urlretrieve(
gh + "/src/data/FeatureFinderCentroided_1_input.mzML", "feature_test.mzML"
)
# Prepare data loading (save memory by only
# loading MS1 spectra into memory)
options = oms.PeakFileOptions()
options.setMSLevels([1])
fh = oms.MzMLFile()
fh.setOptions(options)
# Load data
input_map = oms.MSExperiment()
fh.load("feature_test.mzML", input_map)
input_map.updateRanges()
ff = oms.FeatureFinderAlgorithmPicked()
# Run the feature finder
out_features = oms.FeatureMap() ## our result
seeds = oms.FeatureMap() ## optional: you can provide seeds where FF should take place -- not used here
params = ff.getParameters(); ## we do not modify params for now
ff.run(input_map, out_features, params, seeds)
out_features.setUniqueIds()
fh = oms.FeatureXMLFile()
fh.store("output.featureXML", out_features)
print("Found", out_features.size(), "features")
With a few lines of Python, we are able to run powerful algorithms available in
OpenMS. The resulting data is held in memory (a FeatureMap object) and can be
inspected directly using the help(out_features) comment. It reveals that the
object supports iteration (through the __iter__ function) as well as direct
access (through the __getitem__ function). This means we write code that uses direct access and iteration in
Python as follows:
f0 = out_features[0]
for f in out_features:
print(f.getRT(), f.getMZ())
Each entry in the FeatureMap is a so-called Feature and allows direct
access to the m/z and RT value from Python. Again, we can learn this by
inspecting help(f) or by consulting the manual.
Note: the output file that we have written (output.featureXML) is an
OpenMS-internal XML format for storing features. You can learn more about file
formats in the Reading MS data formats section.
Metabolomics - Untargeted#
For the untargeted detection of small molecule features we can use the FeatureFindingMetabo with prior MassTraceDetection and ElutionPeakDetection.
import pyopenms as oms
from urllib.request import urlretrieve
gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop/doc/pyopenms"
mzML_path = gh + "/src/data/FeatureFinderMetaboIdent_1_input.mzML"
urlretrieve(mzML_path, "ms_data.mzML")
exp = oms.MSExperiment()
oms.MzMLFile().load("ms_data.mzML", exp)
exp.sortSpectra(True)
mass_traces = []
mtd = oms.MassTraceDetection()
mtd_params = mtd.getDefaults()
mtd_params.setValue(
"mass_error_ppm", 5.0
) # set according to your instrument mass error
mtd_params.setValue(
"noise_threshold_int", 3000.0
) # adjust to noise level in your data
mtd.setParameters(mtd_params)
mtd.run(exp, mass_traces, 0)
mass_traces_split = []
mass_traces_final = []
epd = oms.ElutionPeakDetection()
epd_params = epd.getDefaults()
epd_params.setValue("width_filtering", "fixed")
epd.setParameters(epd_params)
epd.detectPeaks(mass_traces, mass_traces_split)
if epd.getParameters().getValue("width_filtering") == "auto":
epd.filterByPeakWidth(mass_traces_split, mass_traces_final)
else:
mass_traces_final = mass_traces_split
fm = oms.FeatureMap()
feat_chrom = []
ffm = oms.FeatureFindingMetabo()
ffm_params = ffm.getDefaults()
ffm_params.setValue("isotope_filtering_model", "none")
ffm_params.setValue(
"remove_single_traces", "true"
) # set false to keep features with only one mass trace
ffm_params.setValue("mz_scoring_by_elements", "false")
ffm_params.setValue("report_convex_hulls", "true")
ffm.setParameters(ffm_params)
ffm.run(mass_traces_final, fm, feat_chrom)
fm.setUniqueIds()
fm.setPrimaryMSRunPath(["ms_data.mzML".encode()])
Metabolomics - Targeted#
FeatureFinderAlgorithmMetaboIdent performs MS1-based targeted feature extraction based on user provided compounds, which are
specified in an assay library (a tab-separated text file). Detected features are stored in a FeatureMap which can be
stored in a FeatureXMLFile. This tool is useful for the targeted extraction of features for a well-defined set of compounds
with known sum formulas and retention times.
For more information on the format of the assay library and available parameters visit the FeatureFinderMetaboIdent documentation.
The pyOpenMS FeatureFinderAlgorithmMetaboIdent needs a list of FeatureFinderMetaboIdentCompound objects as an assay libray for it’s
run() function. We could create that list ourselves or use the following function to read an assay library as .tsv file:
CompoundName |
SumFormula |
Mass |
Charge |
RetentionTime |
RetentionTimeRange |
IsoDistribution |
|---|---|---|---|---|---|---|
2’-O-methylcytidine |
C10H15N3O5 |
0 |
1 |
207.6 |
0 |
0 |
5-formylcytidine |
C10O6N3H13 |
0 |
1 |
269.4 |
0 |
0 |
5-methyluridine |
C10H14N2O6 |
0 |
1 |
291.6 |
0 |
0 |
adenosine |
C10H13N5O4 |
0 |
1 |
220.8 |
0 |
0 |
deoxyadenosine |
C10H13N5O3 |
0 |
1 |
243.0 |
0 |
0 |
inosine |
C10H12N4O5 |
0 |
1 |
264.0 |
0 |
0 |
import csv
# read tsv file and create list of FeatureFinderMetaboIdentCompound
def metaboTableFromFile(path_to_library_file):
metaboTable = []
with open(path_to_library_file, "r") as tsv_file:
tsv_reader = csv.reader(tsv_file, delimiter="\t")
next(tsv_reader) # skip header
for row in tsv_reader:
metaboTable.append(
oms.FeatureFinderMetaboIdentCompound(
row[0], # name
row[1], # sum formula
float(row[2]), # mass
[int(charge) for charge in row[3].split(",")], # charges
[float(rt) for rt in row[4].split(",")], # RTs
[
float(rt_range) for rt_range in row[5].split(",")
], # RT ranges
[
float(iso_distrib) for iso_distrib in row[6].split(",")
], # isotope distributions
)
)
return metaboTable
Now we can use the following code to detect features with FeatureFinderAlgorithmMetaboIdent and store them in a FeatureXMLFile:
from urllib.request import urlretrieve
import pyopenms as oms
gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop/doc/pyopenms"
mzML_path = gh + "/src/data/FeatureFinderMetaboIdent_1_input.mzML"
urlretrieve(mzML_path, "ms_data.mzML")
urlretrieve(
gh + "/src/data/FeatureFinderMetaboIdent_1_input.tsv", "library.tsv"
)
# load ms data from mzML file into MSExperiment
spectra = oms.MSExperiment()
oms.MzMLFile().load("ms_data.mzML", spectra)
# create FeatureFinderAlgorithmMetaboIdent and assign ms data
ff = oms.FeatureFinderAlgorithmMetaboIdent()
ff.setMSData(spectra)
# read library generate a metabo table with compounds
metabo_table = metaboTableFromFile("library.tsv")
# FeatureMap to store results
fm = oms.FeatureMap()
# edit some parameters
params = ff.getParameters()
params[b"extract:mz_window"] = 5.0 # 5 ppm
params[b"extract:rt_window"] = 20.0 # 20 seconds
params[b"detect:peak_width"] = 3.0 # 3 seconds
ff.setParameters(params)
# run the FeatureFinderMetaboIdent with the metabo_table and mzML file path -> store results in fm
ff.run(metabo_table, fm, mzML_path)
# save FeatureMap to file
oms.FeatureXMLFile().store("detected_features.featureXML", fm)
Note: the output file that we have written (detected_features.featureXML) is an
OpenMS-internal XML format for storing features. You can learn more about file
formats in the Reading MS data formats section.
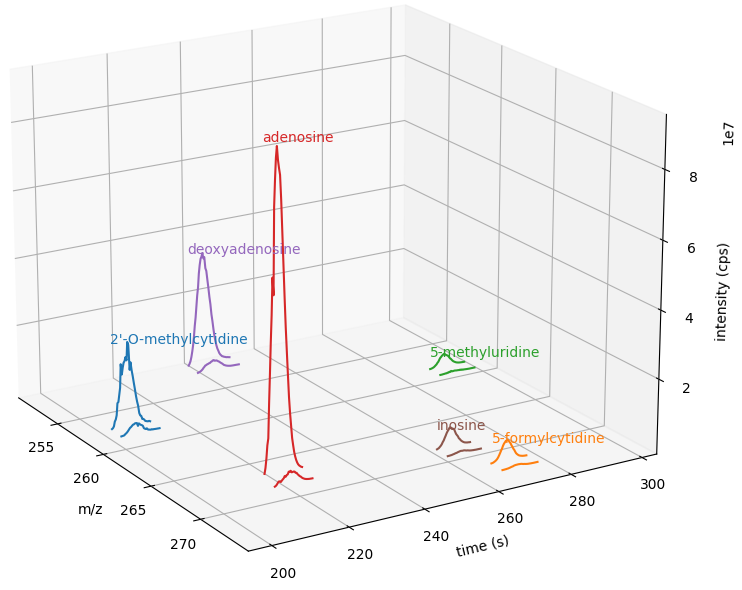
We can get a quick overview on the detected features by plotting them using the following function:
1import matplotlib.pyplot as plt
2import matplotlib.colors as mcolors
3import itertools
4
5def plotDetectedFeatures3D(path_to_featureXML):
6 fm = oms.FeatureMap()
7 fh = oms.FeatureXMLFile()
8 fh.load(path_to_featureXML, fm)
9
10 fig = plt.figure()
11 ax = fig.add_subplot(111, projection="3d")
12
13 cycled_colors = itertools.cycle(['red', 'green', 'blue', 'orange', 'purple', 'yellow', 'cyan', 'magenta', 'black', 'gray'])
14
15 for feature, color in zip(fm, cycled_colors):
16 # chromatogram data is stored in the subordinates of the feature
17 for i, sub in enumerate(feature.getSubordinates()):
18 retention_times = [
19 x[0] for x in sub.getConvexHulls()[0].getHullPoints()
20 ]
21 intensities = [
22 int(y[1]) for y in sub.getConvexHulls()[0].getHullPoints()
23 ]
24 mz = sub.getMZ()
25 ax.plot(retention_times, intensities, zs=mz, zdir="x", color=color)
26 if i == 0:
27 ax.text(
28 mz,
29 retention_times[0],
30 max(intensities) * 1.02,
31 feature.getMetaValue("label"),
32 color=color,
33 )
34
35 ax.set_ylabel("time (s)")
36 ax.set_xlabel("m/z")
37 ax.set_zlabel("intensity (cps)")
38 plt.show()
39
40plotDetectedFeatures3D("detected_features.featureXML")