
Peptides and Proteins#
Sequences of amino acids form peptides and proteins.
Amino Acid Sequences#
The AASequence class handles amino acid sequences in OpenMS. A string of
amino acid residues can be turned into a instance of AASequence to provide
some commonly used operations like chemical formula, weight, and isotope distribution calculations.
The example below shows how amino acid sequences can be created and how basic mass calculations are conducted.
1import pyopenms as oms
2
3seq = oms.AASequence.fromString(
4 "DFPIANGER"
5) # create AASequence object from string representation
6prefix = seq.getPrefix(4) # extract prefix of length 4
7suffix = seq.getSuffix(5) # extract suffix of length 5
8concat = seq + seq # concatenate two sequences
9
10# print string representation of sequences
11print("Sequence:", seq)
12print("Prefix:", prefix)
13print("Suffix:", suffix)
14print("Concatenated:", concat)
15
16# some mass calculations
17mfull = seq.getMonoWeight() # weight of M
18mprecursor = seq.getMonoWeight(oms.Residue.ResidueType.Full, 2) # weight of M+2H
19
20# we can calculate mass-over-charge manually
21mz = seq.getMonoWeight(oms.Residue.ResidueType.Full, 2) / 2.0 # m/z of [M+2H]2+
22# or simply by:
23mz = seq.getMZ(2) # same as above
24
25print()
26print("Monoisotopic mass of peptide [M] is", mfull)
27print("Monoisotopic mass of peptide precursor [M+2H]2+ is", mprecursor)
28print("Monoisotopic m/z of [M+2H]2+ is", mz)
Which prints the amino acid sequence as well as the result of concatenating two sequences or taking the suffix of a sequence. We then compute the mass of the full peptide (\(\ce{[M]}\)), the mass of the peptide precursor (\(\ce{[M+2H]2+}\)) and m/z value of the peptide precursor (\(\ce{[M+2H]2+}\)). Note that, the mass of the peptide precursor is shifted by two protons that are now attached to the molecules as charge carriers. (Detail: the proton mass of \(1.007276\ u\) is slightly different from the mass of an uncharged hydrogen atom at \(1.007825\ u\)). We can easily calculate the charged weight of a \(\ce{(M+2H)2+}\) ion and compute m/z simply dividing by the charge.
Sequence: DFPIANGER
Prefix: DFPI
Suffix: ANGER
Concatenated: DFPIANGERDFPIANGER
Monoisotopic mass of peptide [M] is 1017.4879641373001
Monoisotopic mass of peptide precursor [M+2H]2+ is 1019.5025170708421
Monoisotopic m/z of [M+2H]2+ is 509.7512585354211
The AASequence object also allows iterations directly in Python:
1seq = oms.AASequence.fromString("DFPIANGER")
2
3print("The peptide", str(seq), "consists of the following amino acids:")
4for aa in seq:
5 print(aa.getName(), ":", aa.getMonoWeight())
Which will print
The peptide DFPIANGER consists of the following amino acids:
Aspartate : 133.0375092233
Phenylalanine : 165.0789793509
Proline : 115.0633292871
Isoleucine : 131.0946294147
Alanine : 89.04767922330001
Asparagine : 132.0534932552
Glycine : 75.0320291595
Glutamate : 147.05315928710002
Arginine : 174.1116764466
The N- and C-Terminus as well as the residues themself can be modified. The example below shows how to check for such modifications.
1seq = oms.AASequence.fromString("C[143]PKCK(Label:13C(6)15N(2))CR")
2
3# check if AASequence has a N- or C-terminal modification
4if seq.hasNTerminalModification():
5 print("N-Term Modification: ", seq.getNTerminalModification().getFullId())
6if seq.hasCTerminalModification():
7 print("C-Term Modification: ", seq.getCTerminalModification().getFullId())
8# iterate over all residues and look for modifications
9for aa in seq:
10 if aa.isModified():
11 print(
12 aa.getName(), ":", aa.getMonoWeight(), ":", aa.getModificationName()
13 )
14 else:
15 print(aa.getName(), ":", aa.getMonoWeight())
Which will print:
N-Term Modification: Pyro-carbamidomethyl (N-term C)
Cysteine : 121.01974995329999
Proline : 115.06332928709999
Lysine : 146.1055284466
Cysteine : 121.01974995329999
Lysine : 154.11972844660002 : Label:13C(6)15N(2)
Cysteine : 121.01974995329999
Arginine : 174.1116764466
Molecular Formula#
We can now combine our knowledge of AASequence with what we learned in
about EmpiricalFormula to get accurate mass and isotope distributions from
the amino acid sequence. But first, let’s get the formula of peptide:
1seq = oms.AASequence.fromString("DFPIANGER")
2seq_formula = seq.getFormula()
3print("Peptide", seq, "has molecular formula", seq_formula)
Isotope Patterns#
We now want to print the coarse (e.g., peaks only at nominal masses) distribution.
1# print coarse isotope distribution
2coarse_isotopes = seq_formula.getIsotopeDistribution(
3 oms.CoarseIsotopePatternGenerator(6)
4)
5for iso in coarse_isotopes.getContainer():
6 print(
7 "Isotope", iso.getMZ(), "has abundance", iso.getIntensity() * 100, "%"
8 )
For most applications in computational proteomics, the coarse isotope distribution is sufficient.
But if we deal with very high resolution instruments, we still might want to calculate the isotopic fine structure.
We use the FineIsotopePatternGenerator in OpenMS to reveal these additional peaks:
1# print fine structure of isotope distribution
2fine_isotopes = seq_formula.getIsotopeDistribution(
3 oms.FineIsotopePatternGenerator(0.01)
4) # max 0.01 unexplained probability
5for iso in fine_isotopes.getContainer():
6 print(
7 "Isotope", iso.getMZ(), "has abundance", iso.getIntensity() * 100, "%"
8 )
And plot the very similar looking distributions using standard matplotlib functionality:
1import math
2from matplotlib import pyplot as plt
3
4
5def plotIsotopeDistribution(isotope_distribution, title="Isotope distribution"):
6 plt.title(title)
7 distribution = {"mass": [], "abundance": []}
8 for iso in isotope_distribution.getContainer():
9 distribution["mass"].append(iso.getMZ())
10 distribution["abundance"].append(iso.getIntensity() * 100)
11
12 bars = plt.bar(
13 distribution["mass"], distribution["abundance"], width=0.01, snap=False
14 ) # snap ensures that all bars are rendered
15
16 plt.ylim([0, 110])
17 plt.xticks(
18 range(
19 math.ceil(distribution["mass"][0]) - 2,
20 math.ceil(distribution["mass"][-1]) + 2,
21 )
22 )
23 plt.xlabel("Atomic mass (u)")
24 plt.ylabel("Relative abundance (%)")
25
26
27plt.figure(figsize=(10, 7))
28plt.subplot(1, 2, 1)
29plotIsotopeDistribution(coarse_isotopes, "Isotope distribution - coarse")
30plt.subplot(1, 2, 2)
31plotIsotopeDistribution(fine_isotopes, "Isotope distribution - fine structure")
32plt.show()
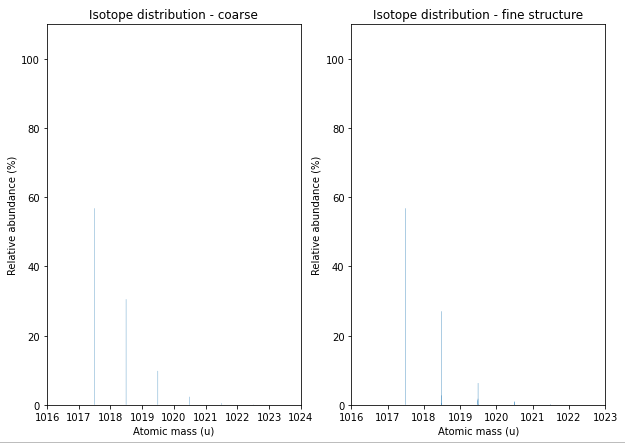
Fragment Ions#
We can easily calculate different ion types for amino acid sequences:
1suffix = seq.getSuffix(3) # y3 ion "GER"
2print("=" * 35)
3print("y3 ion sequence:", suffix)
4y3_formula = suffix.getFormula(oms.Residue.ResidueType.YIon, 2) # y3++ ion
5suffix.getMonoWeight(oms.Residue.ResidueType.YIon, 2) / 2.0 # CORRECT
6suffix.getMonoWeight(oms.Residue.ResidueType.XIon, 2) / 2.0 # CORRECT
7suffix.getMonoWeight(oms.Residue.ResidueType.BIon, 2) / 2.0 # INCORRECT
8
9print("y3 mz:", suffix.getMonoWeight(oms.Residue.ResidueType.YIon, 2) / 2.0)
10print("y3 molecular formula:", y3_formula)
Which will produce
===================================
y3 ion sequence: GER
y3 mz: 181.09514385
y3 molecular formula: C13H24N6O6
Easy, isn’t it? To generate full theoretical spectra watch out for the more specialized
(and faster) TheoreticalSpectrumGenerator which we will take a look at later.
Modified Sequences#
The AASequence class can also handle modifications,
modifications are specified using a unique string identifier present in the
ModificationsDB in round brackets after the modified amino acid or by providing
the mass of the residue in square brackets. For example
AASequence.fromString(".DFPIAM(Oxidation)GER.") creates an instance of the
peptide “DFPIAMGER” with an oxidized methionine. There are multiple ways to specify modifications, and
AASequence.fromString("DFPIAM(UniMod:35)GER"),
AASequence.fromString("DFPIAM[+16]GER") and
AASequence.fromString("DFPIAM[147]GER") are all equivalent.
1 seq = oms.AASequence.fromString("PEPTIDESEKUEM(Oxidation)CER")
2 print(seq.toUnmodifiedString())
3 print(seq.toString())
4 print(seq.toUniModString())
5 print(seq.toBracketString())
6 print(seq.toBracketString(False))
7
8 print(oms.AASequence.fromString("DFPIAM(UniMod:35)GER"))
9 print(oms.AASequence.fromString("DFPIAM[+16]GER"))
10 print(oms.AASequence.fromString("DFPIAM[+15.99]GER"))
11 print(oms.AASequence.fromString("DFPIAM[147]GER"))
12 print(oms.AASequence.fromString("DFPIAM[147.035405]GER"))
The above code outputs:
PEPTIDESEKUEMCER
PEPTIDESEKUEM(Oxidation)CER
PEPTIDESEKUEM(UniMod:35)CER
PEPTIDESEKUEM[147]CER
PEPTIDESEKUEM[147.0354000171]CER
DFPIAM(Oxidation)GER
DFPIAM(Oxidation)GER
DFPIAM(Oxidation)GER
DFPIAM(Oxidation)GER
DFPIAM(Oxidation)GER
Note there is a subtle difference between
AASequence.fromString(".DFPIAM[+16]GER.") and
AASequence.fromString(".DFPIAM[+15.9949]GER.") - while the former will try to
find the first modification matching to a mass difference of \(16 \pm 0.5\), the
latter will try to find the closest matching modification to the exact mass.
The exact mass approach usually gives the intended results while the first
approach may or may not. In all instances, it is better to use an exact description of the desired modification, such as UniMod, instead of mass differences.
N- and C-terminal modifications are represented by brackets to the right of the dots
terminating the sequence. For example, ".(Dimethyl)DFPIAMGER." and
".DFPIAMGER.(Label:18O(2))" represent the labelling of the N- and C-terminus
respectively, but ".DFPIAMGER(Phospho)." will be interpreted as a
phosphorylation of the last arginine at its side chain:
1 s = oms.AASequence.fromString(".(Dimethyl)DFPIAMGER.")
2 print(s, s.hasNTerminalModification())
3 s = oms.AASequence.fromString(".DFPIAMGER.(Label:18O(2))")
4 print(s, s.hasCTerminalModification())
5 s = oms.AASequence.fromString(".DFPIAMGER(Phospho).")
6 print(s, s.hasCTerminalModification())
Arbitrary / unknown amino acids (usually due to an unknown modification) can be
specified using tags preceded by \(\ce{X}\): \(\ce{X[weight]}\). This indicates a new amino
acid (”\(\ce{X}\)”) with the specified weight, e.g. \(\ce{RX[148.5]T}\). Note that this tag
does not alter the amino acids to the left (\(\ce{R}\)) or right (\(\ce{T}\)). Rather, \(\ce{X}\)
represents an amino acid on its own. Be careful when converting such AASequence
objects to an EmpiricalFormula using getFormula(), as tags will not be
considered in this case (there exists no formula for them). However, they have
an influence on getMonoWeight() and getAverageWeight()!
Applying Fixed or Variable Modifications to Sequences#
In this tutorial, we will cover a step-by-step guide on how to use the pyopenms library to generate modified peptides from a given amino acid sequence.
1 import pyopenms as poms
2
3 # Create an amino acid sequence using the fromString() method of the AASequence class.
4 # In this example, we will use the amino acid sequence "TESTMTECSTMTESTR"
5 sequence = poms.AASequence.fromString("TESTMTECSTMTESTR")
6
7 # We use the names "Oxidation (M)" and "Carbamidomethyl (C)" for the variable and fixed modifications, respectively.
8 variable_mod_names = [b"Oxidation (M)"]
9 fixed_mod_names = [b"Carbamidomethyl (C)"]
10
11 # We then use the getModifications() method of the ModifiedPeptideGenerator class to get the modifications for these names.
12 variable_modifications = poms.ModifiedPeptideGenerator.getModifications(variable_mod_names)
13 fixed_modifications = poms.ModifiedPeptideGenerator.getModifications(fixed_mod_names)
14
15 # Apply the fixed modifications to the amino acid sequence
16 poms.ModifiedPeptideGenerator.applyFixedModifications(fixed_modifications, sequence)
17
18 # Define the maximum number of variable modifications allowed
19 max_variable_mods = 1
20
21 # Generate the modified peptides
22 peptides_with_variable_modifications = []
23 keep_unmodified_in_result = False
24 poms.ModifiedPeptideGenerator.applyVariableModifications(variable_modifications, sequence, max_variable_mods,
25 peptides_with_variable_modifications,
26 keep_unmodified_in_result)
27
28 # Print the modified peptides generated using Fixed modifications and their mono-isotopic mass.
29 print("Fixed:", sequence.toString())
30 print("Mono-isotopic mass:", sequence.getMonoWeight())
31
32 # Print the modified peptides generated using variable modifications and their mono-isotopic mass.
33 for peptide in peptides_with_variable_modifications:
34 print("Variable:", peptide.toString())
35 print("Mono-isotopic mass:", peptide.getMonoWeight())
The above code outputs:
Fixed: TESTMTEC(Carbamidomethyl)STMTESTR
Mono-isotopic mass: 1850.7332409542007
Variable: TESTMTEC(Carbamidomethyl)STM(Oxidation)TESTR
Mono-isotopic mass: 1866.7281559542005
Variable: TESTM(Oxidation)TEC(Carbamidomethyl)STMTESTR
Mono-isotopic mass: 1866.7281559542005
Proteins and FASTA Files#
Protein sequences, can be loaded from and stored in FASTA protein databases using FASTAFile.
The example below shows how protein sequences can be stored in FASTA files and loaded back in pyOpenMS:
1 bsa = oms.FASTAEntry() # one entry in a FASTA file
2 bsa.sequence = "MKWVTFISLLLLFSSAYSRGVFRRDTHKSEIAHRFKDLGE"
3 bsa.description = "BSA Bovine Albumin (partial sequence)"
4 bsa.identifier = "BSA"
5 alb = oms.FASTAEntry()
6 alb.sequence = "MKWVTFISLLFLFSSAYSRGVFRRDAHKSEVAHRFKDLGE"
7 alb.description = "ALB Human Albumin (partial sequence)"
8 alb.identifier = "ALB"
9
10 entries = [bsa, alb]
11
12 f = oms.FASTAFile()
13 f.store("example.fasta", entries)
Afterwards, the example.fasta file can be read again from the disk:
1 entries = []
2 f = oms.FASTAFile()
3 f.load("example.fasta", entries)
4 print(len(entries))
5 for e in entries:
6 print(e.identifier, e.sequence)