
Untargeted Metabolomics Pre-Processing#
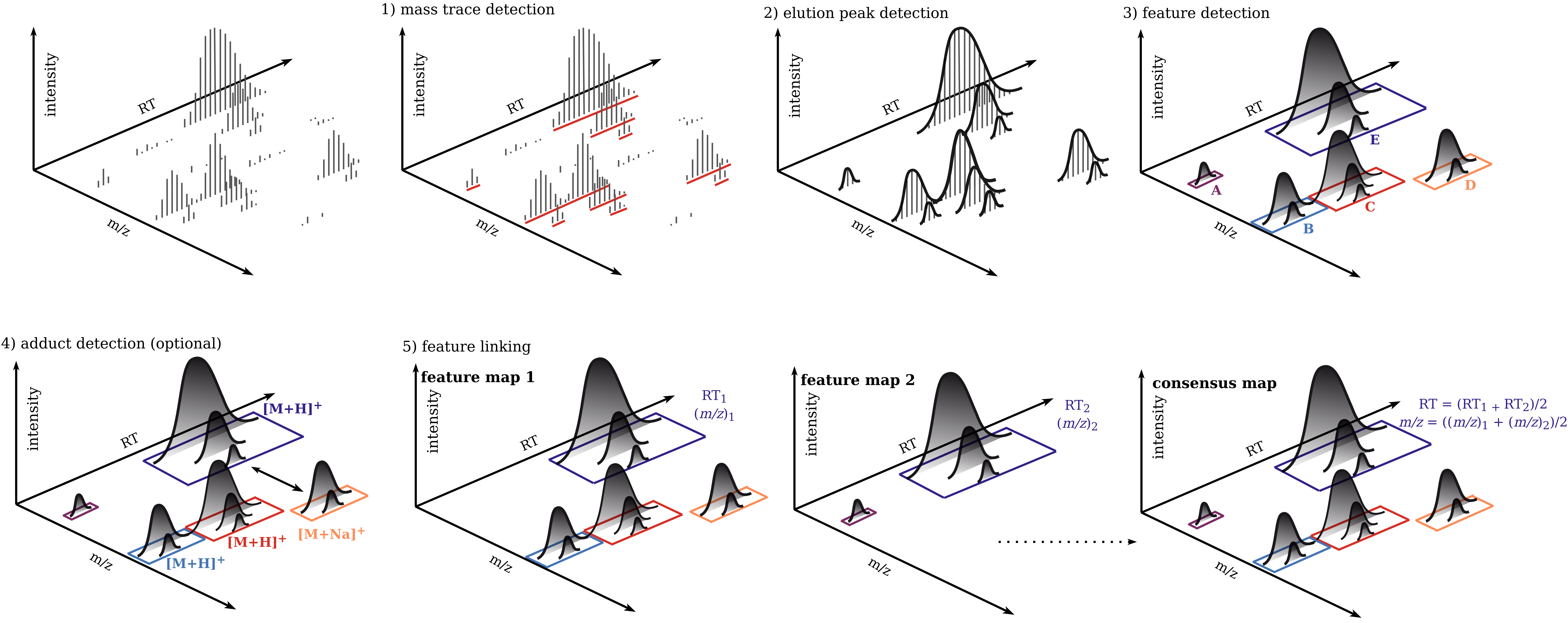
The universal workflow for untargeted metabolomics always consists of feature detection in the individual MS sample files and their linkage to consensus features with common m/z and retention time values. In addition, there are optional steps such as adduct detection and annotation of features with associated MS2 spectra.
First, download two example mzML files.
from urllib.request import urlretrieve
gh = "https://raw.githubusercontent.com/OpenMS/OpenMS/develop/doc/pyopenms"
urlretrieve(gh + "/src/data/Metabolomics_1.mzML", "Metabolomics_1.mzML")
urlretrieve(gh + "/src/data/Metabolomics_2.mzML", "Metabolomics_2.mzML")
For each mzML file do mass trace, elution peak and features detection.
import pyopenms as oms
import os
mzML_files = ["Metabolomics_1.mzML", "Metabolomics_2.mzML"]
feature_maps = []
for file in mzML_files:
# load mzML file into MSExperiment
exp = oms.MSExperiment()
oms.MzMLFile().load(
file, exp
) # load each mzML file to an OpenMS file format (MSExperiment)
# mass trace detection
mass_traces = (
[]
) # introduce an empty list where the mass traces will be loaded
mtd = oms.MassTraceDetection()
mtd_par = (
mtd.getDefaults()
) # get the default parameters in order to edit them
mtd_par.setValue("mass_error_ppm", 10.0) # high-res instrument, orbitraps
mtd_par.setValue(
"noise_threshold_int", 1.0e04
) # data-dependent (usually works for orbitraps)
mtd.setParameters(mtd_par) # set the new parameters
mtd.run(exp, mass_traces, 0) # run mass trace detection
# elution peak detection
mass_traces_deconvol = []
epd = oms.ElutionPeakDetection()
epd_par = epd.getDefaults()
epd_par.setValue(
"width_filtering", "fixed"
) # The fixed setting filters out mass traces outside the [min_fwhm: 1.0, max_fwhm: 60.0] interval
epd.setParameters(epd_par)
epd.detectPeaks(mass_traces, mass_traces_deconvol)
# feature detection
feature_map = oms.FeatureMap() # output features
chrom_out = [] # output chromatograms
ffm = oms.FeatureFindingMetabo()
ffm_par = ffm.getDefaults()
ffm_par.setValue(
"remove_single_traces", "true"
) # remove mass traces without satellite isotopic traces
ffm.setParameters(ffm_par)
ffm.run(mass_traces_deconvol, feature_map, chrom_out)
feature_map.setUniqueIds() # Assigns a new, valid unique id per feature
feature_map.setPrimaryMSRunPath(
[file.encode()]
) # Sets the file path to the primary MS run (usually the mzML file)
feature_maps.append(feature_map)
Align features retention times based on the feature map with the highest number of features (reference map).
1# use as reference for alignment, the file with the largest number of features
2# (works well if you have a pooled QC for example)
3ref_index = feature_maps.index(sorted(feature_maps, key=lambda x: x.size())[-1])
4
5aligner = oms.MapAlignmentAlgorithmPoseClustering()
6
7trafos = {}
8
9# parameter optimization
10aligner_par = aligner.getDefaults()
11aligner_par.setValue("max_num_peaks_considered", -1) # infinite
12aligner_par.setValue(
13 "pairfinder:distance_MZ:max_difference", 10.0
14) # Never pair features with larger m/z distance
15aligner_par.setValue("pairfinder:distance_MZ:unit", "ppm")
16aligner.setParameters(aligner_par)
17aligner.setReference(feature_maps[ref_index])
18
19for feature_map in feature_maps[:ref_index] + feature_maps[ref_index + 1 :]:
20 trafo = oms.TransformationDescription() # save the transformed data points
21 aligner.align(feature_map, trafo)
22 trafos[feature_map.getMetaValue("spectra_data")[0].decode()] = trafo
23 transformer = oms.MapAlignmentTransformer()
24 transformer.transformRetentionTimes(feature_map, trafo, True)
Align mzML files aligment based on FeatureMap alignment (optional, only for GNPS).
1# align mzML files based on FeatureMap alignment and store as mzML files (for GNPS!)
2for file in mzML_files:
3 exp = oms.MSExperiment()
4 oms.MzMLFile().load(file, exp)
5 exp.sortSpectra(True)
6 exp.setMetaValue("mzML_path", file)
7 if file not in trafos.keys():
8 oms.MzMLFile().store(file[:-5] + "_aligned.mzML", exp)
9 continue
10 transformer = oms.MapAlignmentTransformer()
11 trafo_description = trafos[file]
12 transformer.transformRetentionTimes(exp, trafo_description, True)
13 oms.MzMLFile().store(file[:-5] + "_aligned.mzML", exp)
14mzML_files = [file[:-5] + "_aligned.mzML" for file in mzML_files]
Map MS2 spectra to features as PeptideIdentification objects (optional, only for GNPS).
1feature_maps_mapped = []
2use_centroid_rt = False
3use_centroid_mz = True
4mapper = oms.IDMapper()
5for file in mzML_files:
6 exp = oms.MSExperiment()
7 oms.MzMLFile().load(file, exp)
8 for i, feature_map in enumerate(feature_maps):
9 if feature_map.getMetaValue("spectra_data")[
10 0
11 ].decode() == exp.getMetaValue("mzML_path"):
12 peptide_ids = oms.PeptideIdentificationList()
13 protein_ids = []
14 mapper.annotate(
15 feature_map,
16 peptide_ids,
17 protein_ids,
18 use_centroid_rt,
19 use_centroid_mz,
20 exp,
21 )
22 fm_new = oms.FeatureMap(feature_map)
23 fm_new.clear(False)
24 # set unique identifiers to protein and peptide identifications
25 prot_ids = []
26 if len(feature_map.getProteinIdentifications()) > 0:
27 prot_id = feature_map.getProteinIdentifications()[0]
28 prot_id.setIdentifier(f"Identifier_{i}")
29 prot_ids.append(prot_id)
30 fm_new.setProteinIdentifications(prot_ids)
31 for feature in feature_map:
32 pep_ids = oms.PeptideIdentificationList()
33 for pep_id in feature.getPeptideIdentifications():
34 pep_id.setIdentifier(f"Identifier_{i}")
35 pep_ids.push_back(pep_id)
36 feature.setPeptideIdentifications(pep_ids)
37 fm_new.push_back(feature)
38 feature_maps_mapped.append(fm_new)
39feature_maps = feature_maps_mapped
Detect adducts (optional, only for SIRIUS and GNPS Ion Identity Molecular Networking).
1feature_maps_adducts = []
2for feature_map in feature_maps:
3 mfd = oms.MetaboliteFeatureDeconvolution()
4 mdf_par = mfd.getDefaults()
5 mdf_par.setValue(
6 "potential_adducts",
7 [
8 b"H:+:0.4",
9 b"Na:+:0.2",
10 b"NH4:+:0.2",
11 b"H-1O-1:+:0.1",
12 b"H-3O-2:+:0.1",
13 ],
14 )
15 mfd.setParameters(mdf_par)
16 feature_map_adduct = oms.FeatureMap()
17 mfd.compute(feature_map, feature_map_adduct, oms.ConsensusMap(), oms.ConsensusMap())
18 feature_maps_adducts.append(feature_map_adduct)
19feature_maps = feature_maps_adducts
20
21# for SIRIUS store the feature maps as featureXML files!
22for feature_map in feature_maps:
23 oms.FeatureXMLFile().store(
24 feature_map.getMetaValue("spectra_data")[0].decode()[:-4]
25 + "featureXML",
26 feature_map,
27 )
Link features in a ConsensusMap.
1feature_grouper = oms.FeatureGroupingAlgorithmKD()
2
3consensus_map = oms.ConsensusMap()
4file_descriptions = consensus_map.getColumnHeaders()
5
6for i, feature_map in enumerate(feature_maps):
7 file_description = file_descriptions.get(i, oms.ColumnHeader())
8 file_description.filename = os.path.basename(
9 feature_map.getMetaValue("spectra_data")[0].decode()
10 )
11 file_description.size = feature_map.size()
12 file_descriptions[i] = file_description
13
14feature_grouper.group(feature_maps, consensus_map)
15consensus_map.setColumnHeaders(file_descriptions)
16consensus_map.setUniqueIds()
17oms.ConsensusXMLFile().store("FeatureMatrix.consensusXML", consensus_map)
To get a final feature matrix in a table format, export the :consensus features in a pandas DataFrame.
1df = consensus_map.get_df()